
Learning Objectives
- Define cancer as uncontrolled cell division and explain how the normal cell cycle and apoptosis play a role in preventing tumor formation.
- Explain how oncogenes and tumor suppressor genes can mutate to cause cancerous cell division.
- Explain how chemotherapy and radiation can work to treat cancer.
- State the uses of genome sequencing to craft a refined, personalized medicine approach to treating a patient’s cancer.
Cancer and the cell cycle
How cancer can be linked to overactive positive cell cycle regulators (oncogenes) or inactive negative regulators (tumor suppressors). [Modified from Khan Academy. Please link to original article for numbered references.]
Earlier in the semester we learned about cell division and the cell cycle. Does cell cycle control matter? If you ask an oncologist – a doctor who treats cancer patients – she or he will likely answer with a resounding yes.
Cancer is basically a disease of uncontrolled cell division. Its development and progression are usually linked to a series of changes in the activity of cell cycle regulators, the activators that tell the cell to move from G1 to S to G2 to M phases. For example, inhibitors of the cell cycle keep cells from dividing when conditions aren’t right, so too little activity of these inhibitors can promote cancer. Similarly, positive regulators of cell division can lead to cancer if they are too active. In most cases, these changes in activity are due to mutations in the genes that encode cell cycle regulator proteins.
Here, we’ll look in more detail at what’s wrong with cancer cells. We’ll also see how abnormal forms of cell cycle regulators can contribute to cancer.
What’s wrong with cancer cells?
Cancer cells behave differently than normal cells in the body. Many of these differences are related to cell division behavior.
For example, cancer cells can multiply in culture (outside of the body in a dish) without any growth factors, or growth-stimulating protein signals, being added. This is different from normal cells, which need growth factors to grow in culture.
Cancer cells may make their own growth factors, have growth factor pathways that are stuck in the “on” position, or, in the context of the body, even trick neighboring cells into producing growth factors to sustain them [1].
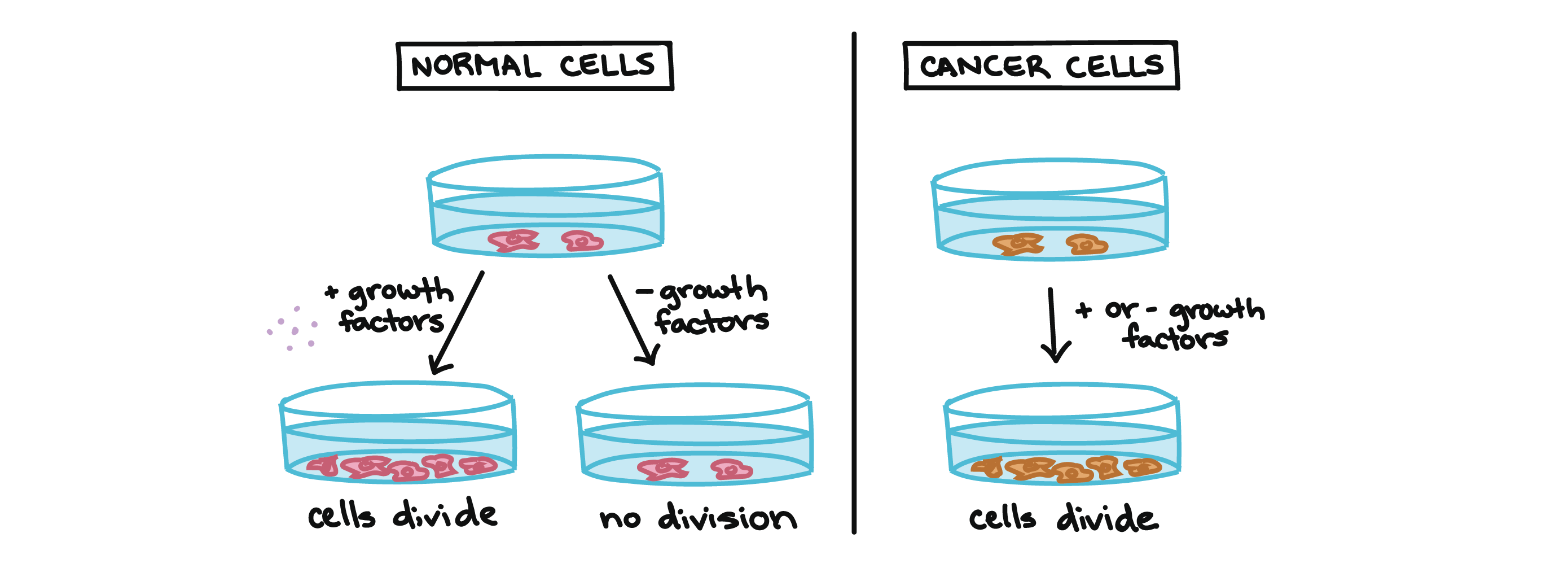
Normal cells in a culture dish will not divide without the addition of growth factors.
-
Cancer cells in a culture dish will divide whether growth factors are provided or not.
Cancer cells also ignore signals that should cause them to stop dividing. For instance, when normal cells grown in a dish are crowded by neighbors on all sides, they will no longer divide. Cancer cells, in contrast, keep dividing and pile on top of each other in lumpy layers.
The environment in a dish is different from the environment in the human body, but scientists think that the loss of contact inhibition in plate-grown cancer cells reflects the loss of a mechanism that normally maintains tissue balance in the body [2].
Another hallmark of cancer cells is their “replicative immortality,” a fancy term for the fact that they can divide many more times than a normal cell of the body. In general, human cells can go through only about 40-60 rounds of division before they lose the capacity to divide, “grow old,” and eventually die [3].
Cancer cells can divide many more times than this, largely because they express an enzyme called telomerase, which reverses the wearing down of chromosome ends that normally happens during each cell division [4].
Cancer cells are also different from normal cells in other ways that aren’t directly cell cycle-related. These differences help them grow, divide, and form tumors. For instance, cancer cells gain the ability to migrate to other parts of the body, a process called metastasis, and to promote growth of new blood vessels, a process called angiogenesis (which gives tumor cells a source of oxygen and nutrients). Cancer cells also fail to undergo programmed cell death, or apoptosis, under conditions when normal cells would (e.g., due to DNA damage). In addition, emerging research shows that cancer cells may undergo metabolic changes that support increased cell growth and division [5].
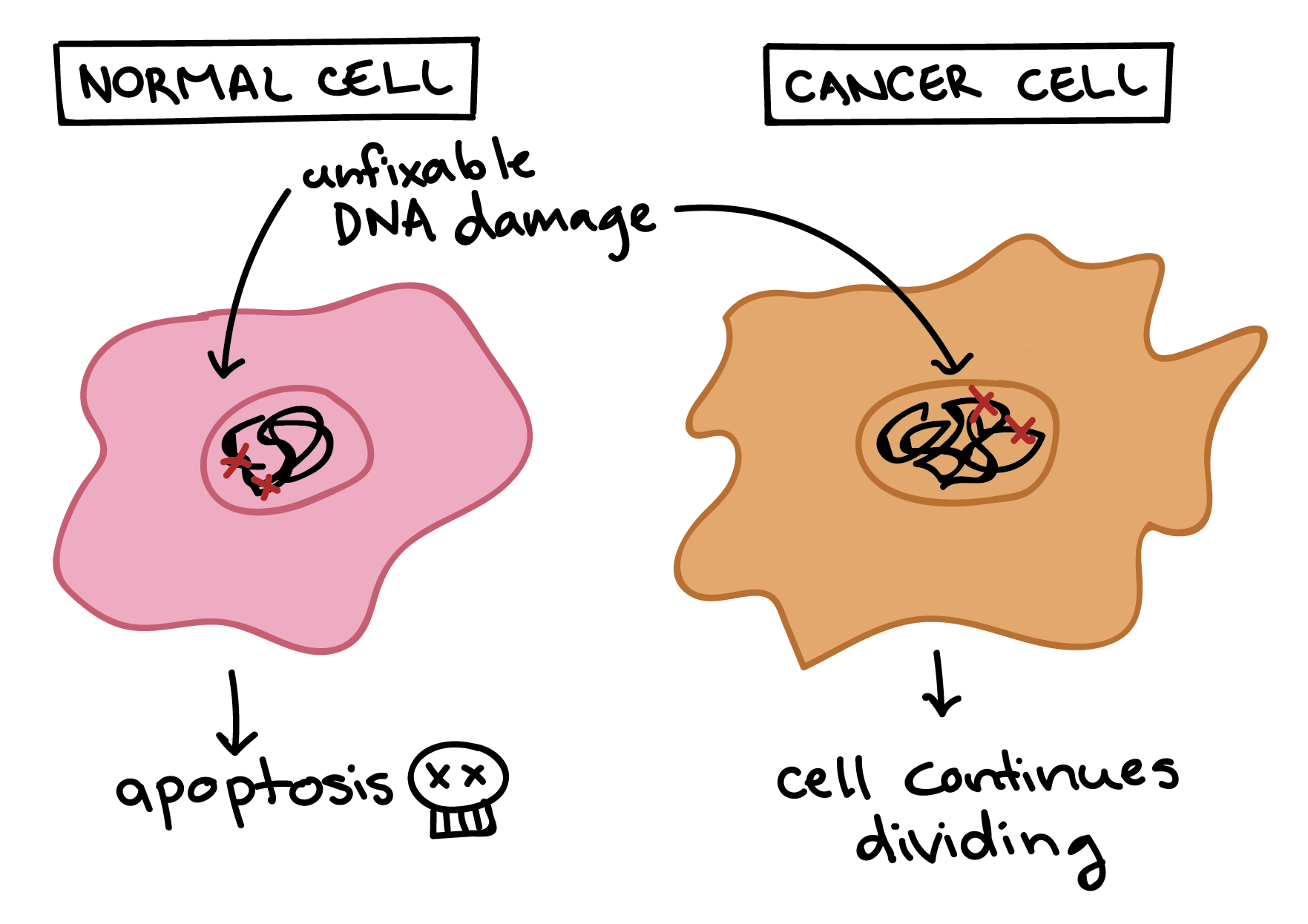
Diagram showing different responses of normal and cancer cells to conditions that would typically trigger apoptosis.
-
A normal cell with unfixable DNA damaged will undergo apoptosis.
-
A cancer cell with unfixable DNA damage will not undergo apoptosis and will instead continue dividing.
How cancer develops
Cells have many different mechanisms to restrict cell division, repair DNA damage, and prevent the development of cancer. Because of this, it’s thought that cancer develops in a multi-step process, in which multiple mechanisms must fail before a critical mass is reached and cells become cancerous. Specifically, most cancers arise as cells acquire a series of mutations (changes in DNA) that make them divide more quickly, escape internal and external controls on division, and avoid programmed cell death [6].
How might this process work? In a hypothetical example, a cell might first lose activity of a cell cycle inhibitor, an event that would make the cell’s descendants divide a little more rapidly. It’s unlikely that they would be cancerous, but they might form a benign tumor, a mass of cells that divide too much but don’t have the potential to invade other tissues (metastasize) [7].
Over time, a mutation might take place in one of the descendant cells, causing increased activity of a positive cell cycle regulator. The mutation might not cause cancer by itself either, but the offspring of this cell would divide even faster, creating a larger pool of cells in which a third mutation could take place. Eventually, one cell might gain enough mutations to take on the characteristics of a cancer cell and give rise to a malignant tumor, a group of cells that divide excessively and can invade other tissues [7].

Diagram of a hypothetical series of mutations that might lead to cancer development.
In the first step, an initial mutation inactivates a negative cell cycle regulator.
In one of the descendants of the original cell, a new mutation takes place, making a positive cell cycle regulator overly active.
In one of the descendants of this second cell, a third mutation takes place, inactivating a genome stability factor.
Once the genome stability factor is inactivated, additional mutations accumulate rapidly in the cell’s descendants (because mutations are no longer prevented or repaired as efficiently).
Once a critical mass of mutations affecting relevant processes is reached, the cell bearing the mutations acquires cancerous characteristics (uncontrolled division, evasion of apoptosis, capacity for metastasis, etc.) and is said to be a cancer cell.
As a tumor progresses, its cells typically acquire more and more mutations. Advanced-stage cancers may have major changes in their genomes, including large-scale mutations such as the loss or duplication of entire chromosomes. How do these changes arise? At least in some cases, they seem to be due to inactivating mutations in the very genes that keep the genome stable (that is, genes that prevent mutations from occurring or being passed on) [8].
These genes encode proteins that sense and repair DNA damage, intercept DNA-binding chemicals, maintain the telomere caps on the ends of chromosomes, and play other key maintenance roles [9]. If one of these genes is mutated and nonfunctional, other mutations can accumulate rapidly. So, if a cell has a nonfunctional genome stability factor, its descendants may reach the critical mass of mutations needed for cancer much faster than normal cells.
Cell cycle regulators and cancer
Different types of cancer involve different types of mutations, and, each individual tumor has a unique set of genetic alterations. In general, however, mutations of two types of cell cycle regulators may promote the development of cancer: positive regulators may be overactivated (become oncogenic), while negative regulators, also called tumor suppressors, may be inactivated.
Oncogenes
Positive cell cycle regulators may be overactive in cancer. For instance, a growth factor receptor may send signals even when growth factors are not there, or a cyclin may be expressed at abnormally high levels. The overactive (cancer-promoting) forms of these genes are called oncogenes, while the normal, not-yet-mutated forms are called proto-oncogenes. This naming system reflects that a normal proto-oncogene can turn into an oncogene if it mutates in a way that increases its activity.
Mutations that turn proto-oncogenes into oncogenes can take different forms. Some change the amino acid sequence of the protein, altering its shape and trapping it in an “always on” state. Others involve amplification, in which a cell gains extra copies of a gene and thus starts making too much protein. In still other cases, an error in DNA repair may attach a proto-oncogene to part of a different gene, producing a “combo” protein with unregulated activity [10].
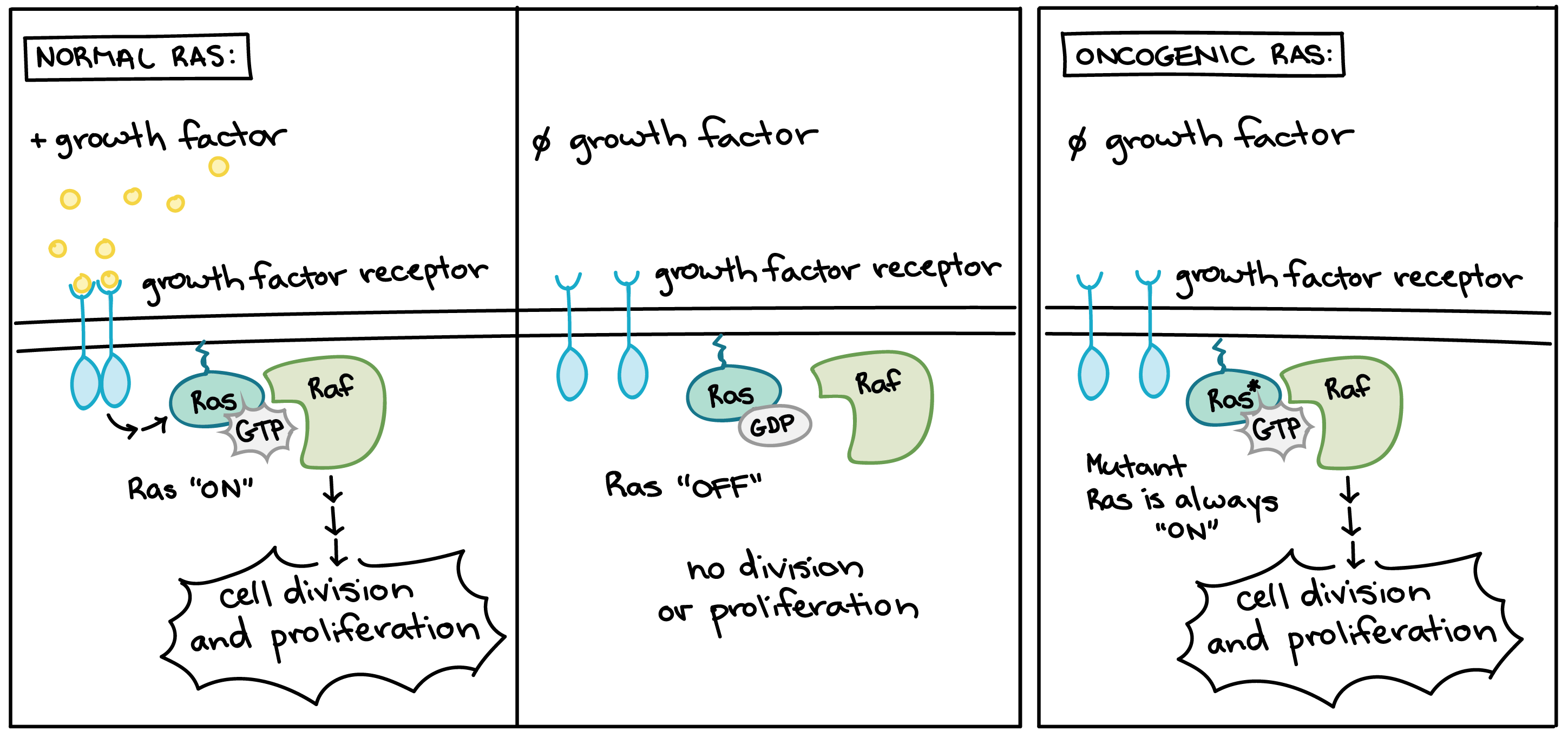
Oncogenic form of the Ras protein.
Normal Ras is activated when growth factors bind to growth factor receptors. When active, Ras switches to its GTP-bound form and triggers a signaling pathway leading to cell division and proliferation. Normal Ras then exchanges GTP for GDP and returns to its inactive state until the cell perceives more growth factors.
An oncogenic form of Ras becomes permanently locked in its GTP-bound, active form. The oncogenic Ras protein activates a signaling pathway leading to growth and proliferation even when growth factors are not present.
Many of the proteins that transmit growth factor signals are encoded by proto-oncogenes. Normally, these proteins drive cell cycle progression only when growth factors are available. If one of the proteins becomes overactive due to mutation, however, it may transmit signals even when no growth factor is around. In the diagram above, the growth factor receptor, the Ras protein, and the signaling enzyme Raf are all encoded by proto-oncogenes [11].
Overactive forms of these proteins are often found in cancer cells. For instance, oncogenic Ras mutations are found in about 90% of pancreatic cancers. Ras is a G protein, meaning that it switches back and forth between an inactive form (bound to the small molecule GDP) and an active form (bound to the similar molecule GTP). Cancer-causing mutations often change Ras’s structure so that it can no longer switch to its inactive form, or can do so only very slowly, leaving the protein stuck in the “on” state (see cartoon above) [12].
Tumor suppressors
Negative regulators of the cell cycle may be less active (or even nonfunctional) in cancer cells. For instance, a protein that halts cell cycle progression in response to DNA damage may no longer sense damage or trigger a response. Genes that normally block cell cycle progression are known as tumor suppressors. Tumor suppressors prevent the formation of cancerous tumors when they are working correctly, and tumors may form when they mutate so they no longer work.
One of the most important tumor suppressors is tumor protein p53, which plays a key role in the cellular response to DNA damage. p53 acts primarily at the G1 checkpoint (controlling the G1 to S transition), where it blocks cell cycle progression in response to damaged DNA and other unfavorable conditions [13].
When a cell’s DNA is damaged, a sensor protein activates p53, which halts the cell cycle at the G1 checkpoint by triggering production of a cell-cycle inhibitor. This pause buys time for DNA repair, which also depends on p53, whose second job is to activate DNA repair enzymes. If the damage is fixed, p53 will release the cell, allowing it to continue through the cell cycle. If the damage is not fixable, p53 will play its third and final role: triggering apoptosis (programmed cell death) so that damaged DNA is not passed on.
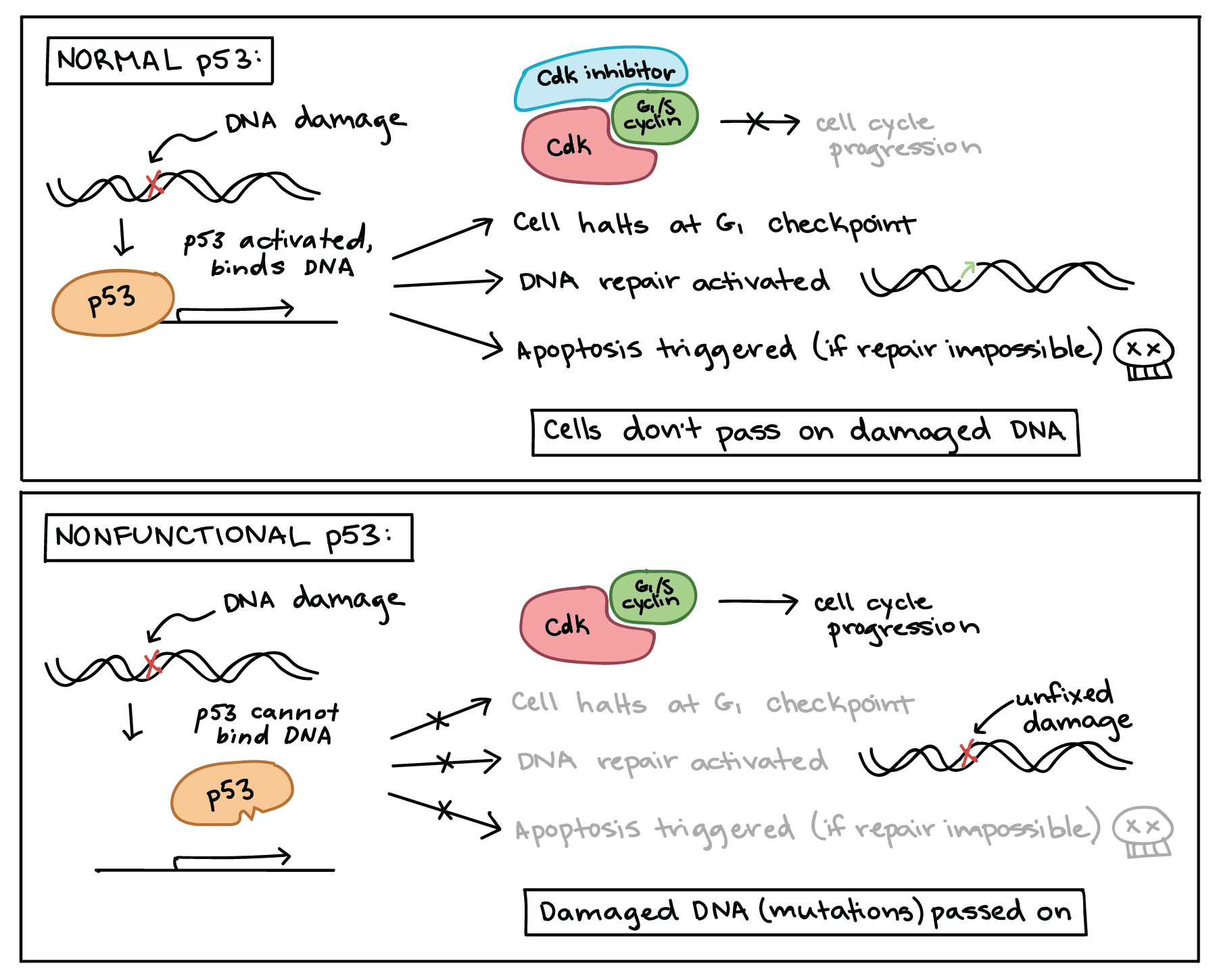
Diagram showing normal p53 and nonfunctional p53.
In response to DNA damage, normal p53 binds DNA and promotes transcription of target genes. First, p53 triggers production of Cdk inhibitor proteins, pausing the cell cycle in G1 to allow time for repairs. p53 also activates DNA repair pathways. Finally, if DNA repair is not possible, p53 triggers apoptosis. The net effect of p53’s activities is to prevent the inheritance of damaged DNA, either by getting the damage repaired or by causing the cell to self-destruct.
When a cell contains only nonfunctional p53 that cannot bind DNA, DNA damage can no longer trigger any of these three responses. Although p53 is still activated by the damage, it is helpless to respond, as it can no longer regulate transcription of its targets. Thus, the cell does not pause in G1, DNA damage is not repaired, and apoptosis is not induced. The net effect of the loss of p53 is to permit damaged DNA (mutations) to be passed on to daughter cells.
In cancer cells, p53 is often missing, nonfunctional, or less active than normal. For example, many cancerous tumors have a mutant form of p53 that can no longer bind DNA. Since p53 acts by binding to target genes and activating their transcription, the non-binding mutant protein is unable to do its job [14].
When p53 is defective, a cell with damaged DNA may proceed with cell division. The daughter cells of such a division are likely to inherit mutations due to the unrepaired DNA of the mother cell. Over generations, cells with faulty p53 tend to accumulate mutations, some of which may turn proto-oncogenes to oncogenes or inactivate other tumor suppressors.
p53 is the gene most commonly mutated in human cancers, and cancer cells without p53 mutations likely inactivate p53 through other mechanisms (e.g., increased activity of the proteins that cause p53 to be recycled) [14,15].
Watch the video below for a summary of oncogenes and tumor suppressors, as well as the current state of cancer treatments, including personalized medicine through genome sequencing:
And wrap up with another perspective from Hank Green on SciShow to get at some of the misconceptions about cancer: