
Learning Objectives
- Define the concept of a fitness trade-off with respect to survival and reproduction, and evaluate a tradeoff graph to explain why organisms cannot simultaneously maximize survival/lifespan and reproduction.
- Define and explain the “mutation accumulation” hypothesis for how the seemingly maladaptive process of aging evolved, and predict the types of genes that could cause aging under the mutation accumulation hypothesis of aging.
- Know that while single gene diseases are rare, they do occur. Explain how a dominant or recessive single gene disease could be inherited, and give some examples of single-gene diseases.
- Define enzyme pathways, explain how gene products can interact in a pathway, and recognize that a single mutation can impact a pathway to cause disease.
- Define polygenic complex trait, recognize polygenic traits by their continuous trait distribution, and read a complex Punnett square to calculate the probability of having offspring for each phenotype.
- Know some examples of disease risk genes, the factors that enhance or decrease risk, and consider the steps you would take to research and learn more about a certain disease if the need arises.
Senescence represents age-related decline in fitness
[Modified from Khan Academy]
Senescence is the age-related decline in an organism’s ability to survive and reproduce. As soon as an organism reach reproductive maturity, it begins to senesce.
All living things need energy and nutrients to grow, maintain their bodies, and reproduce. In nature, these resources are in limited supply, and there is often competition for access to them (e.g., to sunlight and minerals for plants or food sources for animals). Thus, each organism will have non-infinite resources to divide among activities like survival (growth, body maintenance) and reproduction.
What does it mean for an organism to allocate its limited resources “well” in this context? From an evolutionary standpoint, it means that the resources are distributed among the potential activities (growth, maintenance, reproduction) in a way that maximizes fitness, or the relative number of offspring the organism leaves in the next generation. Organisms with inherited traits that cause them to distribute their resources in a more effective way will tend to leave more offspring than organisms lacking these traits, causing the traits to increase in the population over generations by natural selection [2,3].
Over very long periods of time, this process results in species with a collection of life history traits, like number of offspring, timing of reproduction, amount of parental care, etc. These traits are well-adapted for their role and environment. The optimal life history strategy may be different for each species, depending on its traits, environment, and other constraints [2].
That limitation on how energy resources are allocated over a lifetime doesn’t match the perfect world in which an organism would be “immortal and highly fecund,” meaning it would live forever and produce many rounds of offspring, because the more kids you put into the population, the more influence your genes have in future generations (Darwinian fitness). We never see that pattern in nature, and that’s actually a good thing or the earth would be overrun with organisms, and we would have already have run out of resources.
In the real world, organisms experience constraints between growth and reproduction (see figure).
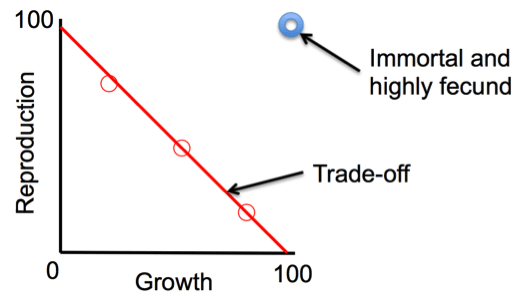
The “ideal” immortal and highly fecund organism does not exist in nature. Instead, the red circles represent trade-offs between high growth, or survival, and high reproduction. Most species fall along the red line. (Image credit: Chrissy Spencer, CC-BY)
The “ideal” immortal and highly fecund organism does not exist in nature. Instead, the red circles represent trade-offs between high growth, or survival, and high reproduction. Most species fall along the red line.
Below, we’ll examine some tradeoffs in life history strategies and see examples of plants and animals that use strategies of different types. For example, plants like coconuts and chestnuts have low fecundity, meaning they produce small numbers of seeds, but each seed is energy-rich and has a good chance of germinating into a new organism. Plants with high fecundity, such as orchids, take the opposite approach: they usually make many small, energy-poor seeds, each of which has a relatively low chance of surviving.
In animals, we also see the tradeoff of many offspring accompanied by low investment/parental care. A typical sea snail (whelk) produces hundreds of eggs at a pop, and these eggs hatch to yield baby snails that are pretty self-sufficient from the get-go. In fact, the baby snails in the first 10 percent of eggs that hatch will enthusiastically eat their slower-hatching siblings for breakfast! [4]
Accumulation of deleterious mutations could explain why we age
These survival and fecundity tradeoffs have been hypothesized to lead to aging! Here’s how that might happen: Imagine you have some genes that are expressed later in life, after you begin reproducing, such as the genes that cause Alzheimer’s or low calcium fixation that increases the risk of bone fractures. If your life history strategy evolves to be live fast, die young, then those later in life genes never become your phenotype because they never get expressed. Mutations that arise in those genes cannot be purged by natural selection acting against them because selection never sees them. Why is that? Selection only acts on phenotypes, not on genotypes, so if a gene is never expressed, its phenotype is never around for selection to act against. In this way, certain genes can accumulate mutations that are never removed from the population by natural selection, so they persist. This mutation accumulation would mean that, should those organisms happen to live a little longer, they will express mutated genes that might cause poor function, disease, or even early death.
Is there any way to halt the aging process?
Halting or even reversing aging would generate immortality, much discussed in fiction and fantasy, and the object of western European explorers in search of the fountain of youth, among others.
Ideas for slowing aging include caloric restriction, enhancing cell-repair, and detoxification mechanisms like enzymes that remove reactive oxygen molecules from cells. We’ll discuss some research ideas on extending lifespan in class.
Here’s a video on senescence in cells, including a few concepts we’ll learn about in the upcoming programmed cell death & cancer reading (telomeres, the Hayflick limit, and apoptosis):
and here’s the SciShow follow up on aging:
Single gene diseases are rare
Remember Punnett squares? We can use them to explain how a single gene disease could be inherited. For example, sickle cell disease occurs when a person has two mutant copies of the hemoglobin gene. Hemoglobin’s normal function in a red blood cell is to bind oxygen for transport to cells throughout the body. A single base change to the genetic code changes one amino acid in the hemoglobin protein. Because of this single mutation, the altered hemoglobin molecules form long fibers that distort the disc-shaped red blood cells into a crescent shape. The cells with crescent or “sickled” shapes cluster and stick in the smallest blood vessels, called capillaries, preventing effective oxygen transport. The sickled blood cells undergo apoptosis more rapidly than normal blood cells. The low oxygen leads to symptoms of anemia, chronic pain, organ damage, and shortened lifespan.
If a disease condition occurs because of loss of function (where a protein stops working due to a mutation in the gene that encodes it), like sickle cell, then having two copies of each gene buffers us against single mutations causing disease. As long as one copy is normal, the normal protein can still carry out its job. However, if both copies become non-functional, then the full condition or disease will manifest. For this reason, most loss of function conditions are usually recessive: alkaptonuria, phenylketonuria, albinism, cystic fibrosis, and sickle cell anemia are all recessive conditions. In some cases, having only one functional copy reduces function, so heterozygotes for, say, sickle cell are often anemic because they don’t circulate sufficient oxygen, but they are not severely debilitated.
Traits are complex, influenced by gene pathways, polygenic inheritance, and environmental effects
[Modified from Khan Academy]
While single gene diseases do exist, they are rare. Most traits are influenced by multiple genes and by the environment in which those genes are expressed, including many characteristics important in our everyday lives, such as height, skin color, eye color, and risk of diseases like diabetes. We’ll take these ideas in turn.
-
Enzyme pathways. An enzymatic pathway is a series of connected chemical reactions that feed one another. The pathway takes in one or more starting molecules and, through a series of intermediates, converts them into products.
In general, each gene encodes a single enzyme that carries out some function in the cell. Enzymes work together to complete tasks in the cell. For instance, a three enzyme pathway is required to convert a starting substrate (green triangle) into the final product needed by the cell (yellow box).
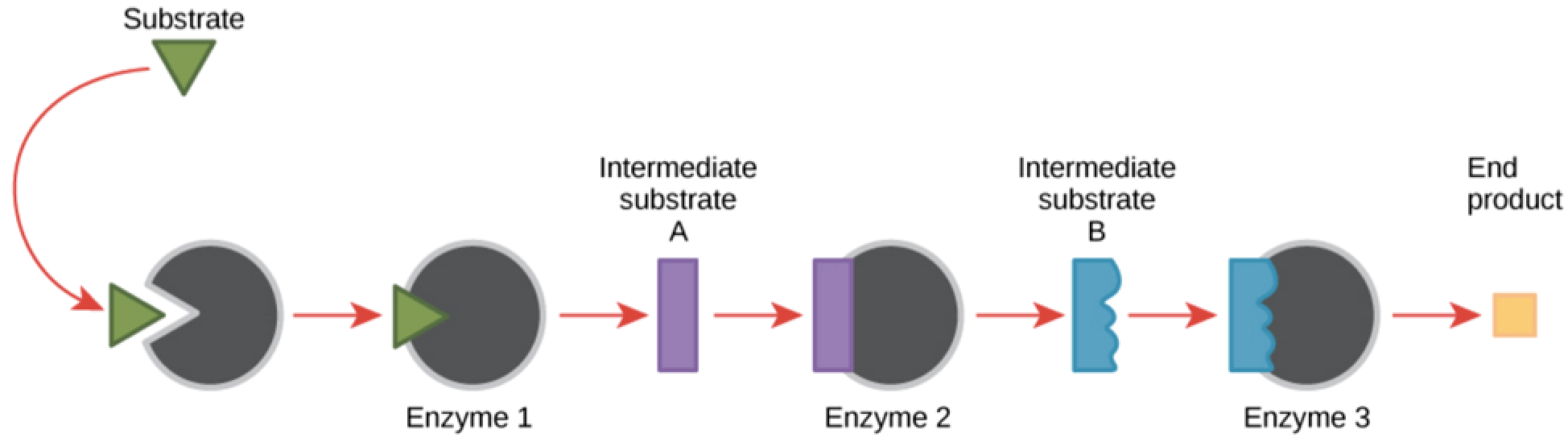
Image credit: OpenStax Biology.
Each enzyme in the pathway converts the substrate into a different intermediate form like an assembly line. If one of those enzymes is missing, the assembly line stops, and the end product needed by the organisms cannot be made. That missing end product could affect the health of the organism, causing disease.
Because each of these enzymes is encoded by a different gene, a single mutation could cause disease by preventing formation of the correct end product. In reality, these pathway traits work pretty much like single gene traits in that if one enzyme’s gene is mutated, the phenotype is affected. Sometimes an intermediate substrate works well enough or is partially functional.
- Polygenic inheritance. Some characteristics are polygenic, meaning that they’re controlled by multiple different genes (not just two). In polygenic inheritance, traits often form a phenotypic spectrum, like heights or pigmentation, rather than falling into clear-cut presence/absence of a trait.
Looking at a real example of a human polygenic trait would get complicated, largely because we’d have to keep track of tens, or even hundreds, of different allele pairs (like the 400 involved in height!). However, we can use an example involving wheat kernels to see how several genes whose alleles “add up” to influence the same trait can produce a spectrum of phenotypes [1,4].
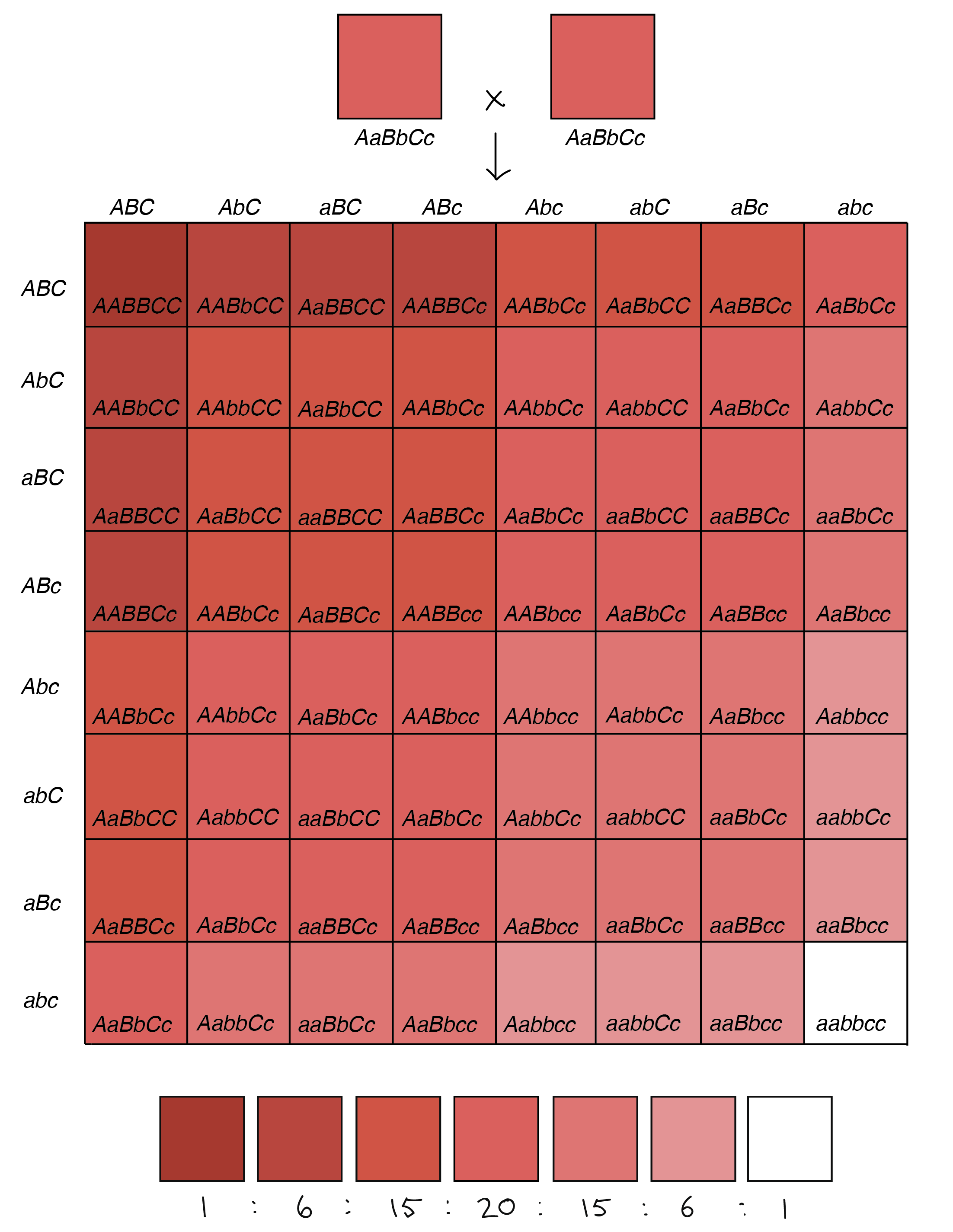
64-square Punnett square illustrating the phenotypes of the offspring of an AaBbCc x AaBbCc cross (in which each uppercase allele contributes one unit of pigment, while each lowercase allele contributes zero units of pigment).
Image credit: Khan Academy. Diagram based on similar diagram by W. P. Armstrong [5].
Now, let’s imagine that two plants heterozygous for all three genes (AaBbCc) were crossed to one another. Each of the parent plants would have three alleles that made pigment, leading to pinkish kernels. Their offspring, however, would fall into seven color groups, ranging from no pigment whatsoever (aabbcc) and white kernels to lots of pigment (AABBCC) and dark red kernels. This is in fact what researchers have seen when crossing certain varieties of wheat [1,4].
This example shows how we can get a spectrum of slightly different phenotypes (something close to continuous variation) with just three genes. It’s not hard to imagine that, as we increased the number of genes involved, we’d be able to get even finer variations in color, or in another trait such as height.
- Environmental effects. Most real-world characteristics are determined not just by genotype, but also by environmental factors that influence how genotype is translated into phenotype.
Reflect back on the sickle cell anemia single-gene disease example. It seems like a simple one-gene disease, but in fact the sickle cell allele conveys different fitnesses in different environments. When the Plasmodium parasites infect and develop in human red blood cells, they cause malaria. But programmed cell death removes the sickled red blood cells from the blood before malaria can develop, so a sickle cell heterozygote tends to have less severe malarial infections in regions where Plasmodium occurs.
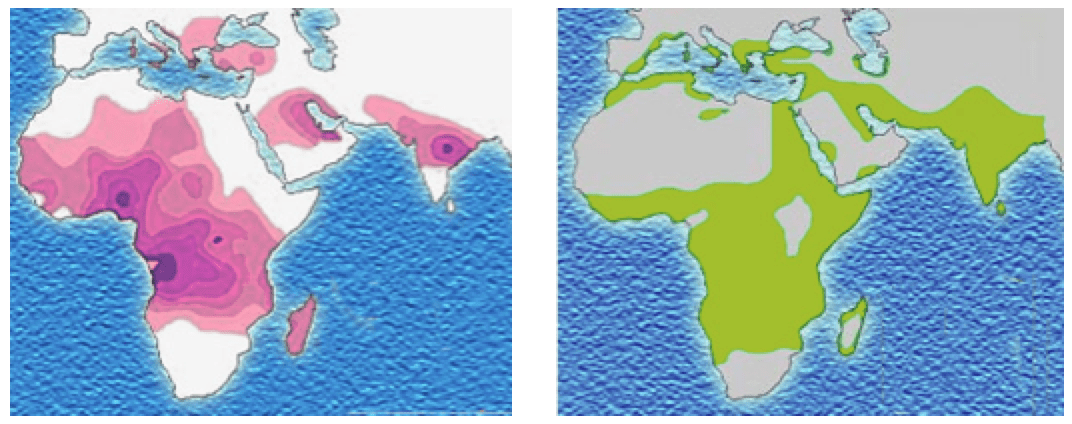
Distribution of the sickle-cell trait shown in pink and purple. Historical distribution of malaria (no longer endemic in Europe) shown in green. (Image credits: Wikipedia CC-LAYOUT; CC-BY-SA-2.5,2.0,1.0)
Variable penetrance, incomplete expressivity
[Modified from Khan Academy]
Some genes enhance disease risk or other traits only under certain conditions, called a gene-by-environment interaction. Even for characteristics that are controlled by a single gene, it’s possible for individuals with the same genotype to have different phenotypes. For example, in the case of a genetic disorder, people with the same disease genotype may have stronger or weaker forms of the disorder, and some may never develop the disorder at all.
In variable expressivity, a phenotype may be stronger or weaker in different people with the same genotype. For instance, in a group of people with a disease-causing genotype, some might develop a severe form of the disorder, while others might have a milder form. The idea of expressivity is illustrated in the diagram below, with the shade of green representing the strength of the phenotype.

The squares in each example are intended to represent individuals of the same genotype for the gene of interest. In narrow expressivity, all six squares are the same. With variable expressivity, the six squares are various shades of green. Illustration modeled after similar image by Steven M. Carr [10]. Image credit: Khan Academy.
The genetic disorder retinoblastoma causes cancerous tumors of the eyes, but the disease varies in severity and speed of onset. Children with retinoblastoma may develop tumors in just one eye or in both eyes, and the tumors may appear more quickly or slowly after birth [9].
In incomplete penetrance, individuals with a certain genotype may or may not develop a phenotype associated with the genotype. For example, among people with the same disease-causing genotype for a hereditary disorder, some might never actually develop the disorder. The idea of penetrance is illustrated in the diagram below, with green or white color representing the presence or absence of a phenotype.
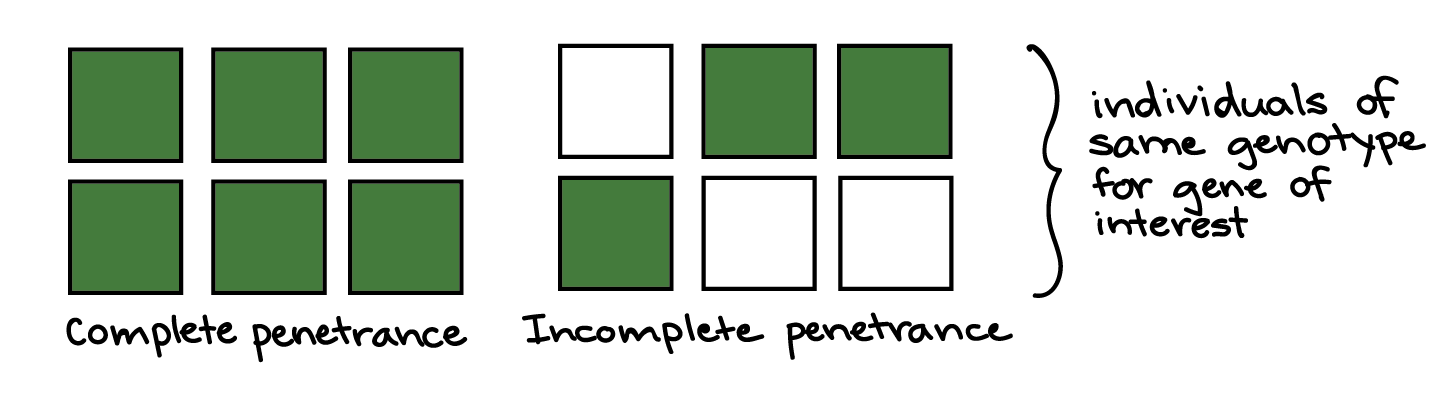
The squares in each example are intended to represent individuals of the same genotype for the gene of interest. With complete penetrance, all six squares are dark green. With incomplete penetrance, three of the squares are dark green, and three of the squares are white. Illustration modeled after similar image by Steven M. Carr [10]. Image credit: Khan Academy.
What causes variable expressivity and incomplete penetrance? Other genes and environmental effects are often part of the explanation. For example, disease-causing alleles of one gene may be suppressed by alleles of another gene elsewhere in the genome, or a person’s overall health may influence the strength of a disease phenotype [11].
Predicting Disease Risk at the Individual Level
[Modified from Openstax Biology]
Currently, we can predict the risk of certain diseases by screening the genomes of healthy individuals. Medical practitioners can recommend lifestyle changes and drugs before disease onset. As you might imagine, genomic screening works best for diseases and conditions caused by single gene defects. But such defects only account for approximately 5 percent of diseases in developed countries. Most of the common diseases, such as heart disease, are polygenic and also involve environmental factors such as diet. In April 2010, scientists at Stanford University published the genome analysis of a healthy individual (Stephen Quake, a scientist at Stanford University). The researchers analyzed Quake’s risk probability for 55 different medical conditions. He learned he had a rare genetic mutation that carried a risk for sudden heart attack. He also has a 23% chance of developing prostate cancer and a 1.4% chance of developing Alzheimer’s. Even though genomic sequencing is becoming more affordable and reliable each year, our society has yet to tackle the ethical issues surrounding genomic analysis at a population level.